
Islam, M.; Yang, Y.; Simmons, A.J.; Shah, V.M.; Musale, K.P.; Xu, Y.; Tasneem, N.; Chen, Z.; Trinh, L.T.; Molina, P.; Ramirez-Solano, M.A.; Sadien, I.D.; Dou, J.; Rolong, A.; Chen, K.; Magnuson, M.A.; Rathmell, J.C.; Macara, I.G.; Winton, D.J.; Liu, Q.; Zafar, H.; Kalhor, R.; Church, G.M.; Shrubsole, M.J.; Coffey, R.J.; Lau, K.S. “Temporal recording of mammalian development and precancer.” Nature, Volume 634, Issue 8036, 2024, pp. 1187-1195, Article 8, DOI: 10.1038/s41586-024-07954-4.
Understanding when and how cells change over time is crucial for studying biology. Traditionally, this involves continuous observation, but another method uses permanent genetic changes, like mutations, as “timestamps” to track events after they happen. Researchers developed a “molecular clock” using CRISPR technology to record the timing of cell changes along with information about cell type and lineage (family tree). This approach revealed the timing of specific cell growth during mouse development, unexpected connections between different cell types, and new states of epithelial cells based on their genetic history.
The method was also applied to study the early stages of colon cancer in mice and humans. By analyzing 418 human precancerous polyps, researchers discovered that 15–30% originated from multiple normal cells rather than a single cell. This innovative framework combines genetic “timestamps” with single-cell analysis, providing new insights into the timing and origins of development and diseases like cancer.
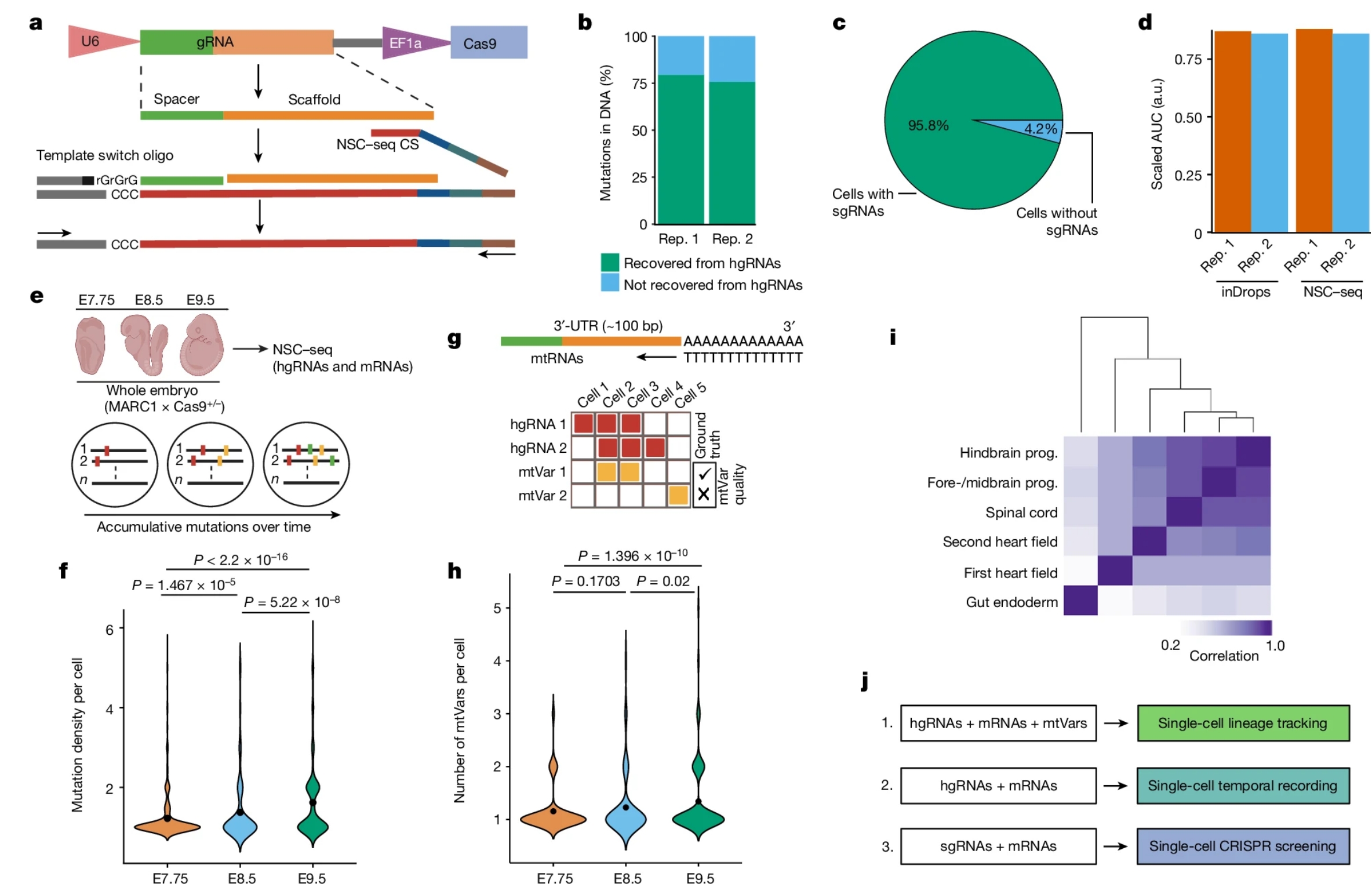
Fig. 1: Optimization of a multipurpose, single-cell capture platform.
a, gRNA capture schematic for the NSC–seq platform. The target site of gRNA scaffold anneals to NSC–seq capture sequence (CS) with a cellular barcode (blue) and unique molecular identifier (green). An additional sequence (grey) is added to the 3′-end of the complementary DNA via template switching during reverse transcription to enable downstream library amplification. This gRNA capture approach is compatible with any type of gRNA (single-guide RNA (sgRNA), hgRNA and self-targeting guide RNA) that contains the target site sequence in the scaffold (Extended Data Fig. 1). b, Cas9-induced mutation recovery by direct hgRNA capture as compared with mutations detected in DNA of the same samples. c, gRNA capture efficiency by NSC–seq assessed in an experiment in which all cells from a drug-selected cell line should contain sgRNAs. d, Comparative transcriptome capture efficiency between standard inDrops and NSC–seq experiments. e, NSC–seq experiments performed on developmentally barcoded whole embryos in which Cas9 is constitutively expressed (top). Accumulative mutations on homing barcode regions increase over time (bottom)5,20. f, Average mutation density over embryonic time points (Extended Data Fig. 2a). Black dots represent geometric mean for each time point, and P values are derived from unpaired two-tailed t-tests. g, Somatic mtVar calling from mitochondrial RNA (mtRNA) (top). Approach to filtering informative mtVars for lineage tracking using hgRNA mutations as ground truth (bottom) (Extended Data Fig. 3b–d). h, Number of somatic mtVars per cell over embryonic time points. Black dot represents geometric mean for each time point, and P values were derived from unpaired two-tailed t-tests. i, Pearson correlation coefficient heat map of variant proportions combining hgRNAs and mtVars for selected tissue types, presented as pseudobulk from an E9.5 embryo (Extended Data Fig. 4). j, Multimodal application of the NSC–seq platform. a,e,g,j, Schematics created using BioRender (https://BioRender.com). a.u., arbitrary units; AUC, area under the curve; rep., replicate; prog., progenitor; bp, base pairs.