Teasing out a role for tepsin in AP-4 mediated trafficking
 AP-4 is one complex in a family of coat proteins that mediate vesicle formation at various membranes. Previous work has determined that AP-4 is recruited specifically to the trans-Golgi network, but the exact mechanisms governing AP-4 vesicle budding or distribution are still poorly understood. Further, loss of the AP-4 complex in humans results in a rare disorder termed AP-4 deficiency syndrome. In this work, members of the Jackson lab, including postdoctoral fellows Dr. Natalie Wallace and Dr. John Gadbery, graduate student Cameron Cohen and lab manager Amy Kendall, a combination of biochemical and imaging approaches are used to investigate tepsin, an AP-4 accessory protein with an unknown function.
AP-4 is one complex in a family of coat proteins that mediate vesicle formation at various membranes. Previous work has determined that AP-4 is recruited specifically to the trans-Golgi network, but the exact mechanisms governing AP-4 vesicle budding or distribution are still poorly understood. Further, loss of the AP-4 complex in humans results in a rare disorder termed AP-4 deficiency syndrome. In this work, members of the Jackson lab, including postdoctoral fellows Dr. Natalie Wallace and Dr. John Gadbery, graduate student Cameron Cohen and lab manager Amy Kendall, a combination of biochemical and imaging approaches are used to investigate tepsin, an AP-4 accessory protein with an unknown function.
Through a series of pulldowns and isothermal titration calorimetry measurements, tepsin was shown to bind LC3B. LC3B is a protein central to autophagy, a process by which autophagosomes break down damaged or diseased proteins/organelles. During the development of autophagosomes, LC3B becomes conjugated to the autophagosome membrane and recruits cargo through LC3B-interacting (LIR) motifs. Accordingly, tepsin was found to contain a LIR motif which binds the LIR-interacting site of LC3B.
Interestingly, the tepsin-LC3B interaction is not the first connection between AP-4 and autophagy. ATG9A, an AP-4 cargo, is a lipid scramblase responsible for the even distribution of lipids in the growing autophagosome membrane. Tepsin was 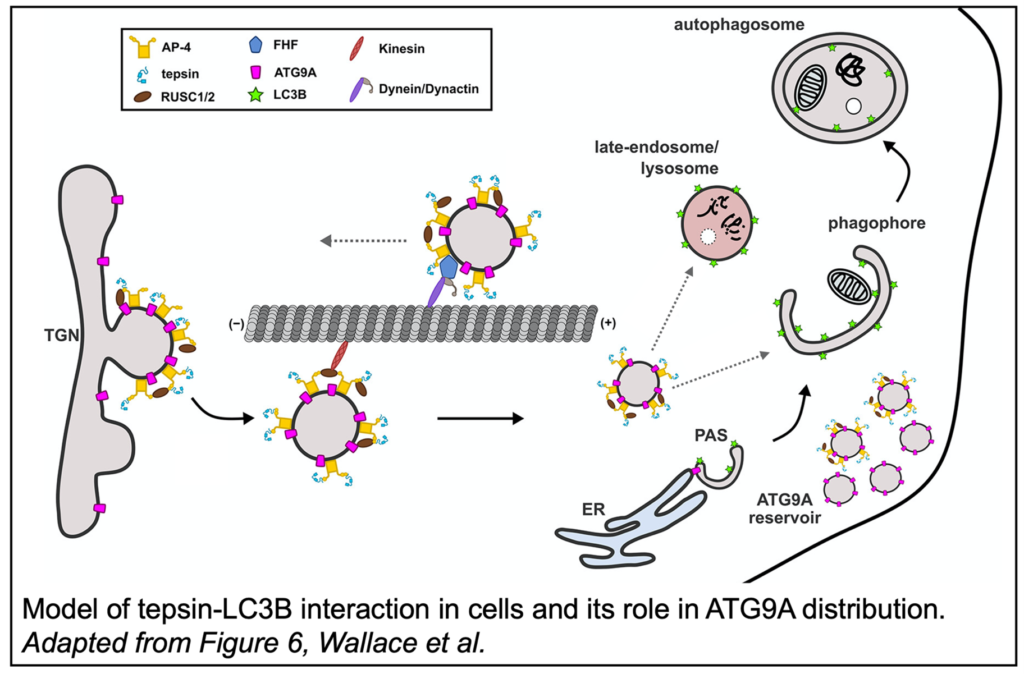 knocked down in HeLa cells and the distribution of ATG9A was assessed via fluorescence microscopy. In the absence of tepsin, ATG9A vesicles were notably redistributed to the cell periphery and the colocalization of ATG9A and LC3B was reduced. Further, tepsin with a mutated LIR motif was reintroduced to these tepsin-depleted cells and failed to restore normal ATG9A distribution, demonstrating the specific importance of tepsin-LC3B binding on ATG9A trafficking. Tepsin depletion was also quantified in HeLa cells with an mRFP-GFP-LC3B reporter, a fusion protein which emits different fluorescent signals depending on the maturity of the autophagosomes to which they are conjugated. Loss of tepsin was shown to result in dysregulated autophagosomes and autolysosomes, the fusion product of autophagosomes and lysosomes.
knocked down in HeLa cells and the distribution of ATG9A was assessed via fluorescence microscopy. In the absence of tepsin, ATG9A vesicles were notably redistributed to the cell periphery and the colocalization of ATG9A and LC3B was reduced. Further, tepsin with a mutated LIR motif was reintroduced to these tepsin-depleted cells and failed to restore normal ATG9A distribution, demonstrating the specific importance of tepsin-LC3B binding on ATG9A trafficking. Tepsin depletion was also quantified in HeLa cells with an mRFP-GFP-LC3B reporter, a fusion protein which emits different fluorescent signals depending on the maturity of the autophagosomes to which they are conjugated. Loss of tepsin was shown to result in dysregulated autophagosomes and autolysosomes, the fusion product of autophagosomes and lysosomes.
Overall, this work has characterized a novel interaction between tepsin and LC3B which is shown to modulate AP-4 dependent ATG9A trafficking. Furthermore, this work solidifies the ties between AP-4 biology and autophagy and increases our understanding of the molecular biology underpinning AP-4 deficiency syndrome.
Read more about this exciting work published in Molecular Biology of the Cell! ~ Cameron I. Cohen
Leave a Response
You must be logged in to post a comment