Unmasking antagonists: a deep dive into the structural binding poses of PPARγ ligands

Peroxisome proliferator-activated receptor γ (PPARγ) is a ligand-regulated transcription factor located in the nucleus. Endogenous ligands, such as lipids and fatty acids, bind an orthosteric pocket in the ligand-binding domain (LBD) of PPARγ and function as agonists to activate gene expression related to adipogenesis and insulin sensitization. Synthetic small molecule ligands, including FDA-approved antidiabetic drugs, also serve as agonists by binding to the same pocket.
In the absence of ligand, PPARγ cycles between active and repressive conformations. Agonist binding to the orthosteric pocket stabilizes the active conformation, specifically by stabilizing the solvent exposed conformation of helix 12 in the LBD, enabling high-affinity binding of coactivators and increasing transcription. GW9662 and T0070907 are synthetic covalent ligands, originally described as antagonists, which slow the rate of exchange between transcriptionally active and repressive conformations and block binding of certain PPARγ agonists.
While it was previously thought that covalent and synthetic ligands would always compete for binding to the PPARγ orthosteric pocket, recent work has indicated that the two ligands can sometimes cobind. However, the structural basis and mechanism of cobinding remains unclear. In this study, Doug Kojetin, associate professor of biochemistry, and a collaborator from the Guangzhou Laboratory, use NMR and crystallography to elucidate the binding pose of GW9662 and T0070907 cobound with synthetic ligands for the first time. 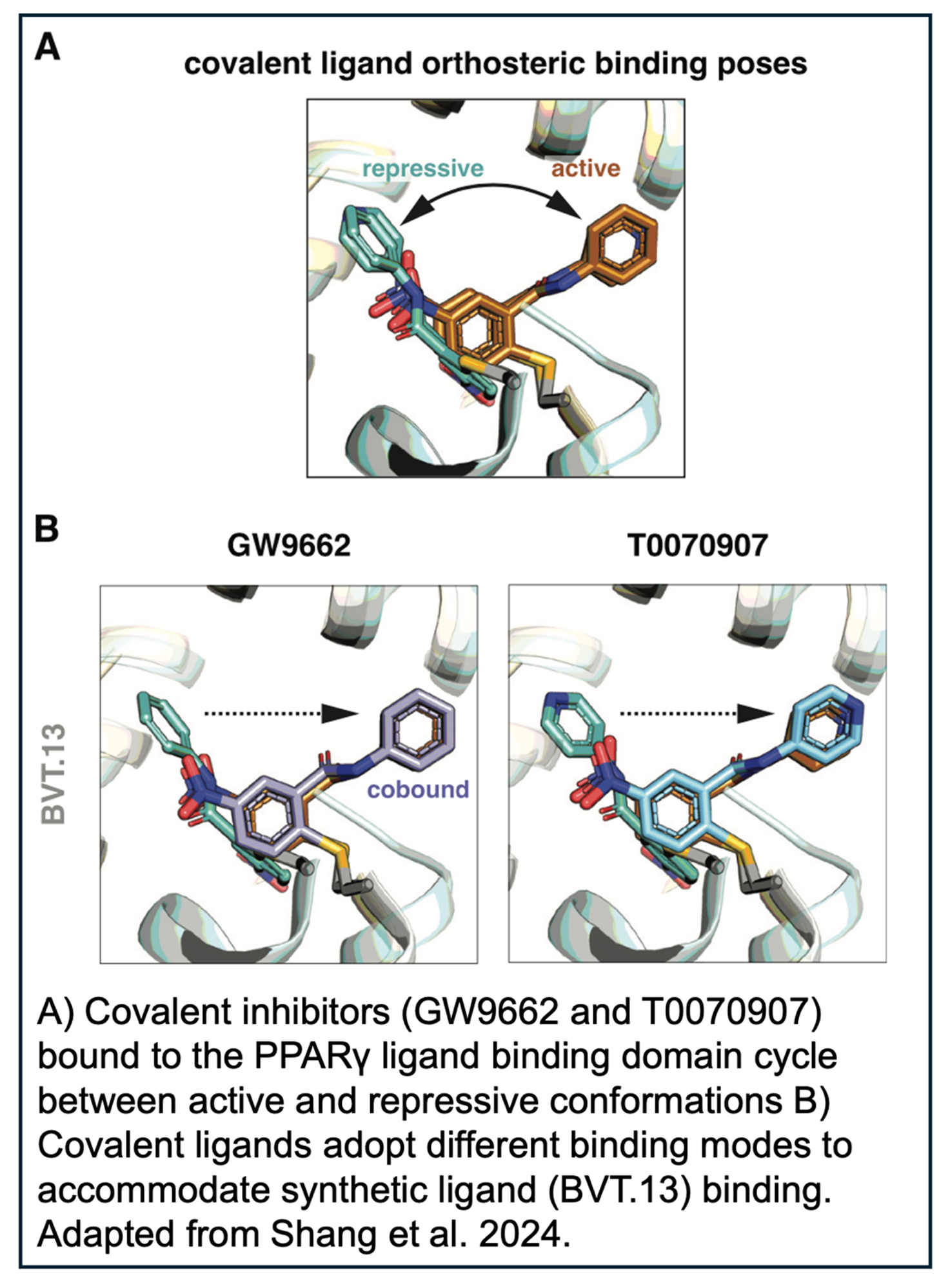
Before diving into structure, the authors used time-resolved fluorescence energy transfer (TR-FRET) to determine how four non-covalent synthetic ligands, BVT-13, MRL24, nTZDpa and SR1664, affected PPARγ activity in the presence of covalent inhibitors. With either covalent inhibitor, GW9662 or T0070907, bound to the LBD of PPARγ, increased affinity between the LBD and a corepressor peptide was observed. Titration in of the synthetic agonists decreased the affinity of the corepressor peptide, indicating that the covalent inhibitors were not sufficient to block agonist binding.
NMR footprinting of the cobound LBD demonstrated that synthetic agonists displace helix 12 in the covalent inhibitor bound orthosteric pocket from a repressive to a solvent-exposed active conformation. In order to obtain more structural detail, the authors then crystalized complexes of PPARγ LBD covalently bound to GW9662 or T0070907 and soaked in the synthetic agonists.
Overall, the LBD conformations were very similar and most of the structures showed that the synthetic ligands bound via their native binding modes or a slightly adapted pose by pushing the covalent ligand aside. While each synthetic agonist utilized unique mechanisms to cobind with the covalent ligands, it is clear that GW9662 or T0070907 are not sufficient to block agonist activity. In the future, it will be necessary to use alternate covalent inhibitors, such as SR16832, which was developed by the authors, to test new PPARγ ligands.
Check out the full article in eLife to get all the cobinding structural details! ~ Cameron I. Cohen
Leave a Response
You must be logged in to post a comment