Putting the fun back in antifungals: new insights into Acanthamoeba drug targets

Acanthamoeba is a genus of free-living pathogenic protozoa found ubiquitously in the environment. In humans, acanthamoeba can cause diseases such as blinding keratitis, an infection of the eye, or granulomatous amebic encephalitis (GAE), a generally fatal infection of the brain and spinal cord. Antifungals which target the biosynthesis of fungal sterols, such as ergosterol, are often used as treatment, but the Acanthamoeba ergosterol biosynthetic pathway has been shown to differ significantly from that found in fungi. It is therefore vital to characterize Acanthamoeba ergosterol biosynthesis and determine the effectiveness of current antifungal compounds.
Azole drugs, a common class of antifungals, block ergosterol synthesis by inhibiting essential cytochrome p450 enzyme sterol 14alpha-demethylase (CYP51) of the fungal sterol pathway. Acanthamoeba castellanii, a pathogenic member of the Acanthamoeba genus, has a CYP51 enzyme but the effect of azoles on its activity has yet to be determined.
In this study, published in the Journal of Medicinal Chemistry, Galina Lepesheva, research professor in biochemistry, and collaborators use a combination of structural, spectral and activity-based assays to characterize A. castellanii CYP51.
The researchers first conducted a sequence analysis of A. castellanii CYP51 which revealed that this protein is much more closely related to plants and algae than yeast and fungi. Further, time course studies of the activity of CYP51 demonstrated significantly higher catalytic efficiency for obtusifoliol over lanosterol as a substrate, supporting an ergosterol biosynthesis pathway analogous to the one in plants. Obtusifoliol was also 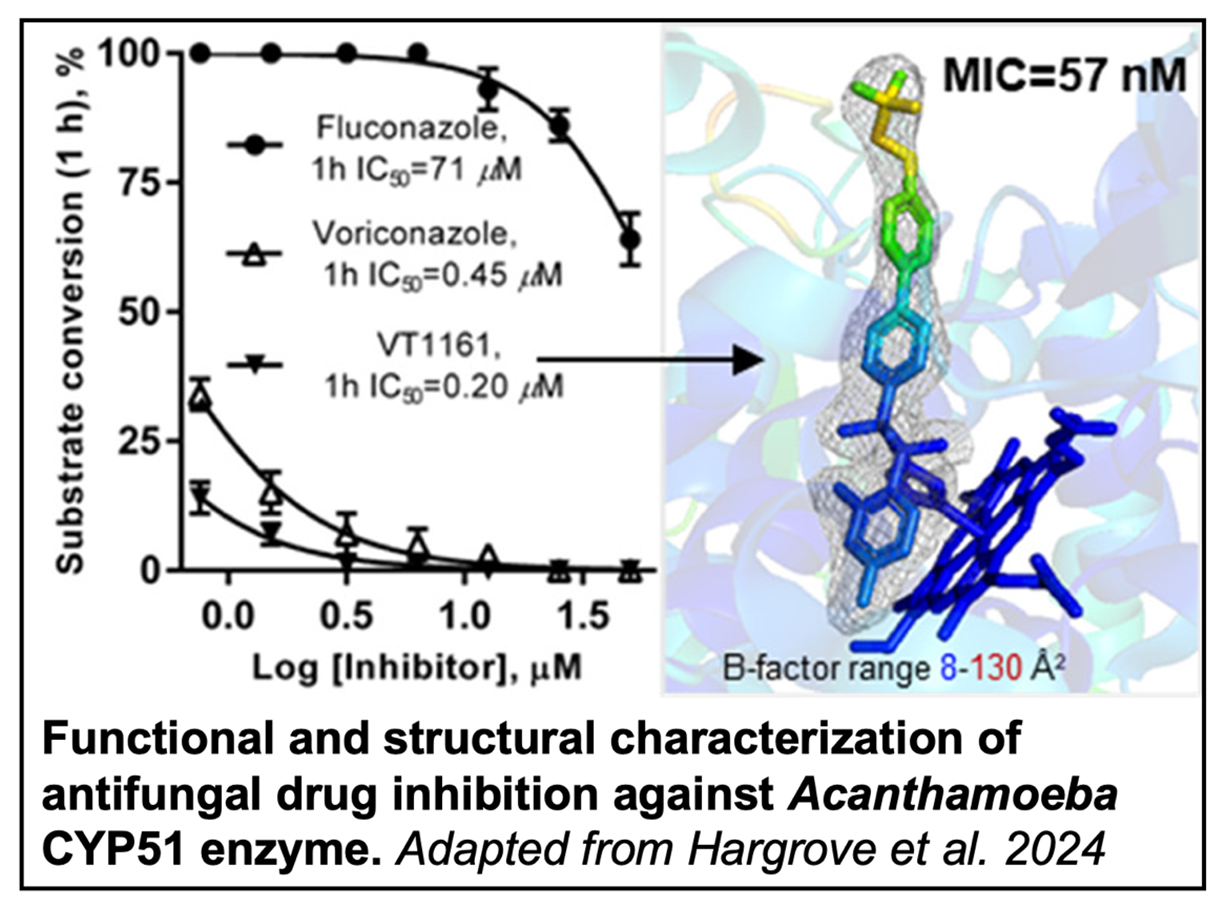 shown to bind with 40-fold higher affinity than lanosterol when assayed by spectral titrations.
shown to bind with 40-fold higher affinity than lanosterol when assayed by spectral titrations.
The efficacy of several antifungal drugs against A. castellanii were then assessed by a CYP51 enzyme activity assay and a cellular growth inhibition assay. Tetrazole-based oteseconazole (VT1161) displayed the most potent inhibition in both cases. Truncated CYP51 protein was subsequently crystalized in the presence and absence of VT1161. Due to the lack of large conformational changes between the two structures, the authors believe the potency of VT1161 is a result of the drug-enzyme interaction preventing required conformational changes for catalysis.
As a newly approved clinical drug, VT1161 is an exciting prospect for the treatment of Acanthamoeba infections. Additionally, the ability of VT1161 to penetrate the blood-brain barrier and its inability to bind human CYP51 make it a promising scaffold for the rational design of new selective inhibitors. Further, the structural and functional information gleaned about the A. castellanii CYP51 enzyme will also inform future drug design.
Read more about this interesting amoeba here! ~ Cameron I. Cohen
Leave a Response
You must be logged in to post a comment